If you use genetics to differentiate populations, the new package smartsnp might be your new friend. Written in R language and available from GitHub and CRAN, this package does principal component analysis with control for genetic drift, projects ancient samples onto modern genetic space, and tests for population differences in genotypes. The package has been built to load big datasets and run complex stats in the blink of an eye, and is fully described in a paper published in Methods in Ecology and Evolution (1).

In the bioinformatics era, sequencing a genome has never been so straightforward. No surprise that > 20 petabytes of genomic data are expected to be generated every year by the end of this decade (2) — if 1 byte of information was 1 mm long, we could make 29,000 round trips to the moon with 20 petabytes. Data size in genetics keeps outpacing the computer power available to handle it at any given time (3). Many will be familiar with a computer freezing if unable to load or run an analysis on a huge dataset, and how many coffees or teas we might have drunk, or computer screens might have been broken, during the wait. The bottom line is that software advances that speed up data processing and genetic analysis are always good news.
With that idea in mind, I have just published a paper presenting the new R package smartsnp (1) to run multivariate analysis of big genotype data, with applications to studies of ancestry, evolution, forensics, lineages, and overall population genetics. I am proud to say that the development of the package has been one of the most gratifying short-term collaborations in my entire career, with my colleagues Christian Huber and Ray Tobler: a true team effort!
The package is available on GitHub and the Comprehensive R Archive Network CRAN. See downloading options here, and vignettes here with step-by-step instructions to run different functionalities of our package (summarised below).
In this blog, I use “genotype” meaning the combination of gene variants (alleles) across a predefined set of positions (loci) in the genome of a given individual of animal, human, microbe, or plant. One type of those variants is single nucleotide polymorphisms (SNP), a DNA locus at which two or more alternative nucleotides occur, sometimes conditioning protein translation or gene expression. SNPs are relatively stable over time and are routinely used to identify individuals and ancestors in humans and wildlife.
What the package does
The package smartsnp is partly based on the field-standard software EIGENSOFT (4, 5) which is only available for Unix command-line environments. In fact, our driving motivation was (i) to broaden the use of EIGENSOFT tools by making them available to the rocketing community of professionals, not only academics who employ R for their work (6), and (ii) to optimise our package to handle big datasets and complex stats efficiently. Our package mimics EIGENSOFT’s principal component analysis (SMARTPCA) (5), and also runs multivariate tests for population differences in genotypes as follows:
- Showing genetic relationships in a simple graph: If you have sequenced multiple SNPs from a range of study individuals (belonging to different populations), PCA can display the genetic resemblance among individuals in a scatter diagram (known as ordination). The points in the ordination are the individuals, and the closer the individuals are in ordination space, the more similar their genotypes are, and vice versa. The package smartsnp implements arguably the two most popular tools of SMARTPCA: (i) prior to analysis, SNP variation across individuals can be scaled to control for the expected allele-frequency dispersion caused by genetic drift (5); and (ii) posterior to analysis, individuals for which some SNPs could not be sequenced (missing nucleotides) can be projected onto the PCA space (7). The latter projection will be useful to compare genotypes for ancient versus modern individuals (see one of our vignettes explaining how to do it with real ancient-human data), or as an alternative to imputation for individuals resulting in low-coverage DNA with lots of missing bases (8).
- Quantifying statistical support for genetic relationships: Our package also implements two statistical tests based on permutations (normality not required). Each study individual must be assigned to one of at least two groups, and those groups might reflect, say, different populations, geographical regions or times. PERMANOVA (9) tests for differences in ‘location’ (populations clustered in different locations of the PCA space because each population has a unique genotype), and PERMDISP (10) tests for differences in ‘dispersion’ (populations show different spread in PCA space because the individuals of some populations have more variable [technically: heteroscedastic] genotypes than those of other populations).
How the package does it
The package smartsnp has four functions — one computes SMARTPCA, PERMANOVA and PERMDISP in a single go (smart_mva), and the three stand-alone functions compute only one analyses separately (smart_pca, smart_permanova, smart_permdisp).
For instance, once the package has been downloaded in your computer, if you set the path to your working directory in the R environment, store your genotype data (my_data) and group vector (my_groups) in the working directory, and run the next line of code:
smart_mva(snp_data = mydata, sample_group = my_groups)
… then you are computing SMARTPCA, PERMANOVA and PERMDISP at once for the genotypes in mydata given the assignment of individuals to groups specified in my_groups.
The function has multiple arguments but the above code executes the default options (e.g., missing values coded 9 in mydata, SNPs with missing values imputed with means, all SNPS scaled to control for genetic drift, PCA calculation truncated to principal axes one and two to accelerate computations, 9,999 permutations and Holm’s correction for multiple pairwise testing).
The function smart_mva will spit out (among other outputs) the position of individuals in PCA space (which you can then plot in R or elsewhere), and the F statistic and p values for PERMANOVA and PERMDISP.
In our paper, we show that smart_pca is 2 to 4 times faster than EIGENSOFT’s SMARTPCA across a range of data sizes. All four functions complete analyses on datasets with up to 100 samples and 1 million SNPs in < 30 seconds, and accurately recreate previously published SMARTPCA on genotypes of wolves Canis lupus (11) and modern and ancient humans Homo sapiens (12).
Genotype data formats
The package smartsnp has been conceived for the analysis of biallelic SNPs with genotypes [0|1|2] from diploid organisms. Thus, a SNP with reference allele G and variant allele T will have genotypes g(GG)= 0 (homozygous reference), g(GT) = 1 (heterozygous) and g(TT) = 2 (homozygous non-reference). Genotypes from haploid or polyploid organisms can be similarly defined and used in our package.
We tailored our package to detect automatically the format of the input data. smartsnp accepts a general flat (text) file or a compressed or uncompressed EIGENSOFT file. Those formats are stable and should preserve our package’s functionality over time (so not requiring constant updates).
In contrast, genetic data formats such as VCF and PLINK are complex and change frequently (13), implying a range of conceptual and programmatic subtleties (14). Nevertheless, we have created a vignette (based on the software plink2) guiding users on transforming the VCF and PLINK/bed formats into a genotype text format (*.traw) accepted by smartsnp. Alternatively, users can resort to other R tools for data conversion into formats accepted by our package (e.g., genomic_converter, glactools, SNPRelate).
Stats pointers
You might be familiar with the following issues, but a reminder seems appropriate here:
- a priori hypothesis: the assignment of individuals to groups must be done according to a hypothesis drawn from theory before doing any analysis. Our package smartsnp allows the combination of PCA with PERMANOVA and PERMDISP based on those a priori expectations. Otherwise, assigning individuals to groups based on a visual inspection of a PCA would be flawed, like testing for differences in the colour between two groups of birds by eyeballing their plumage rather than by formulating some type of prediction (e.g., feather colour might be expected to be driven by different diets or habitats).
- p values: ANOVA hypothesis testing (as in PERMANOVA, PERMDISP) invariably results in a probability (p) of the observed differences between groups if the null hypothesis (zero differences) was true. The probability is informative but does not prove anything and depends on sample size (the higher the sample size, the higher the likelihood that a p value will be low). Accordingly, avoid statements like “… there were significant differences between groups” as they have no statistical or biological meaning, but do state why the magnitude of those differences is biologically relevant (15).
- mean versus variance: ANOVA tests might result in a low p because, for any given measured variable, the mean or the variance or both differ between groups. To tell any of the latter scenarios, an ANOVA must be combined with a homoscedasticity (variance) test. The terms ‘mean’ and ‘variance’ in the univariate case (1 variable as in a one-way ANOVA) equate to ‘location’ and ‘dispersion’ in the multivariate case (> 1 variable as in PCA). Notably, PERMANOVA is rather robust to dispersion effects for balanced designs (16).
Across the board, homoscedasticity should not be seen as nuisance that makes simple ANOVA complicated (this impression mostly results from widespread narratives presenting homogeneity of variance as an assumption of ANOVA tests) but as a statistical property of the data that can have genuine biological meaning.
Thus, in ecology, multivariate dispersion can be interpreted as a measure of (i) species turnover across species assemblages (17), and (ii) stress in species assemblages exposed to environmental perturbations (18). Akin to those interpretations, increased genetic heterogeneity in PCA space for human data can signal (i) increased genetic diversity when the most variable loci have the strongest effects on the phenotype of certain ethnic groups (19-21), or (ii) indicate disease profiles and their ancestral origin (22-25).
Example with real data
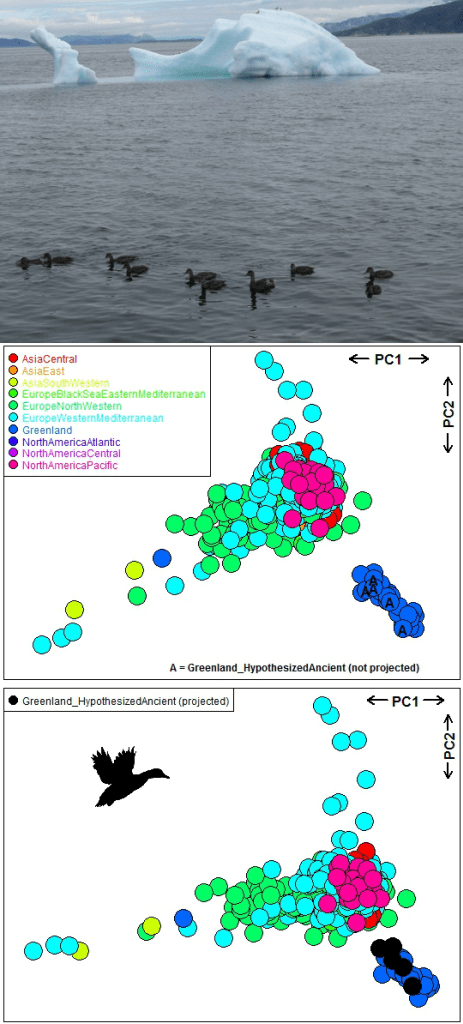
Kraus and colleagues (26) examined the global flyways of mallards Anas platyrhynchos using 364 SNPs (all SNPs had missing values) from 695 individuals belonging to 55 populations and 10 flyways. This study supports panmixia (random mating) or lack of genetic differentiation among flyways except for the Greenland flyway, suggesting
“… the world’s mallards, perhaps with minor exceptions, form a single large, mainly interbreeding population”.
(26)
Plotting smart_pca outputs clearly shows that Greenland individuals cluster in the lower-left corner of the PCA 1 × PCA 2 ordination, while individuals from the other flyways are mixed (see plot below). We have written a vignette showing how to run PCA, PERMANOVA and PERMDISP on this mallard dataset using our package’s four functions.
Our package smartsnp should make SMARTPCA available to a much wider audience than currently, and also enhance the exposition of the genetic concepts behind this numerical method (5).
The functionality of our package should have attractive properties for the growing community of scientists investigating modern and ancient population structure in humans and other taxa and across a range of disciplines (particularly archaeology, conservation biology, (palaeo)ecology, human/wildlife forensics, genetics/genomics, palaeontology, and population and evolutionary biology).
Christian Huber is the maintainer of the package. We are happy to address any issues and questions that smartsnp users might raise, with the bonus that those interactions are bound to improve and expand future versions of the package.
Christian and I were funded by ARC DECRA Fellowship (DE180100883) to him. Ray was funded by ARC DECRA Fellowship (DE190101069). I was supported by the ARC Centre of Excellence for Australian Biodiversity and Heritage (CE170100015), and the EU LIFE programme (LIFE18 NAT/ES/000121 DIVAQUA). We thank Julia Pilowsky, Simon Tuke and Joshua Schmidt for guidance for package editing for GitHub/CRAN, Robert Maier for sharing his code for handling genetic-data formats, and Małgorzata Pilot, Wiesław Bogdanowicz, and Robert H. S. Kraus for sharing their data and assisting statistical interpretations.
References
- S. Herrando-Pérez, R. Tobler, C. D. Huber, smartsnp, an R package for fast multivariate analyses of big genomic data. Meth Ecol Evol in doi:10.1111/2041-210X.13684 (2021).
- F. C. P. Navarro et al., Genomics and data science: an application within an umbrella. Genome Biol 20, 109 (2019).
- V. Marx, The big challenges of big data. Nature 498, 255-260 (2013).
- A. L. Price et al., Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904-909 (2006).
- N. Patterson, A. L. Price, D. Reich, Population structure and eigenanalysis. PLoS Gen 2, e190 (2006).
- S. Tippmann, Programming tools: adventures with R. Nature 517, 109-110 (2014).
- P. R. C. Nelson, P. A. Taylor, J. F. MacGregor, Missing data methods in PCA and PLS: score calculations with incomplete observations. Chemometrics Intellig Lab Syst 35, 45-65 (1996).
- J. Marchini, B. Howie, Genotype imputation for genome-wide association studies. Nat Rev Gen 11, 499-511 (2010).
- M. J. Anderson, A new method for non-parametric multivariate analysis of variance. Austral Ecol 26, 32-46 (2001).
- M. J. Anderson, Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62, 245-253 (2006).
- M. Pilot et al., Global phylogeographic and admixture patterns in grey wolves and genetic legacy of an ancient Siberian lineage. Sci Rep 9, 17328 (2019).
- I. Lazaridis et al., Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419-424 (2016).
- C. C. Chang et al., Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
- H. Zhang, in Statistical Genomics: Methods and Protocols, E. Mathé, S. Davis, Eds. (Springer New York, New York, NY, 2016), pp. 3-17.
- D. H. Johnson, The insignificance of statistical significance testing. J Wildl Manage 63, 763-772 (1999).
- M. J. Anderson, D. C. I. Walsh, PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol Monogr 83, 557-574 (2013).
- M. J. Anderson, K. E. Ellingsen, B. H. McArdle, Multivariate dispersion as a measure of beta diversity. Ecol Lett 9, 683-693 (2006).
- R. M. Warwick, K. R. Clarke, Increased variability as a symptom of stress in marine communities. J Exp Mar Biol Ecol 172, 215-226 (1993).
- N. Solovieff et al., Clustering by genetic ancestry using genome-wide SNP data. BMC Gen 11, 108 (2010).
- C. Yu, G. Ni, J. van der Werf, S. H. Lee, Detecting genotype-population interaction effects by ancestry principal components. Front Gen 11, 379 (2020).
- K. Fadhlaoui-Zid et al., Sousse: extreme genetic heterogeneity in North Africa. J Hum Gen 60, 41-49 (2015).
- A. Manichaikul et al., Population structure of hispanics in the United States: the multi-ethnic study of atherosclerosis. PLoS Gen. 8, e1002640 (2012).
- S. Turajlic, A. Sottoriva, T. Graham, C. Swanton, Resolving genetic heterogeneity in cancer. Nature Reviews Genetics 20, 404-416 (2019).
- J. P. A. Ioannidis, E. E. Ntzani, T. A. Trikalinos, ‘Racial’ differences in genetic effects for complex diseases. Nat Gen 36, 1312-1318 (2004).
- H. N. Horne et al., Fine-mapping of the 1p11.2 breast cancer susceptibility locus. PLoS One 11, e0160316 (2016).
- R. H. S. Kraus et al., Global lack of flyway structure in a cosmopolitan bird revealed by a genome wide survey of single nucleotide polymorphisms. Mol Ecol 22, 41-55 (2013).





Leave a comment